

New technology for protein complex discovery holds promise for biotechnology and crop improvement
Living cells survive and adapt by forming stable protein complexes that allow them to modulate protein activity, do mechanical work and convert signals into predictable responses, but identifying the proteins in those complexes is technically challenging. Purdue University researchers have developed a method to predict the composition of thousands of proteins complexes at one time, a discovery that will speed discoveries about cell functions.
The method predicts the composition of naturally occurring protein complexes that are extracted from living cells. It is significantly faster and cheaper than traditional methods that use large-scale cloning, affinity labels or antibodies to identify protein complex components. The method has the potential to help scientists understand how thousands of protein complexes function together to enable plant cells to grow normally and respond to changing environments.
Daniel Szymanski, a professor in Purdue’s Department of Botany and Plant Pathology, and graduate students Zach McBride and Youngwoo Lee, separated thousands of proteins based on size and charge and used mass spectrometry to predict which proteins were likely to bind to one another and form a stable protein complex. In this guilt-by-association approach, proteins that form a stable complex should co-purify with each other using any separation strategy.
Szymanski’s team also validated the process. The team confirmed the presence of many known and novel protein complexes that were predicted from the profiling method.
“From one of these separations, we get elution profiles for thousands of proteins,” said Szymanski, whose findings were published in the journal Molecular and Cellular Proteomics. “We can combine all of the protein profile data from the columns, identify the elution profiles that are most similar to each other, and predict which proteins are physically associated with one another.”
Once protein complexes are identified, scientists can determine their function in cells, how cellular pathways are regulated, how those proteins affect cell signaling, and more. Szymanski said the method works in any organism that has a sequenced genome, including corn, soybeans, rice, and cotton.
“This method has been used to globally analyze protein complexes in plants of differing genotypes or those grown under different conditions. It’s like a new phenotyping tool to analyze systems-level changes in protein abundance, binding partners, and subcellular localization,” Szymanski said.
The method serves as a large-scale hypothesis-generating machine that will accelerate understanding of the complicated workings of plant cells and give researchers broad knowledge about how plants adapt to heat, water, and other stresses.
The research was supported by the National Science Foundation Plant Genome Research Program.
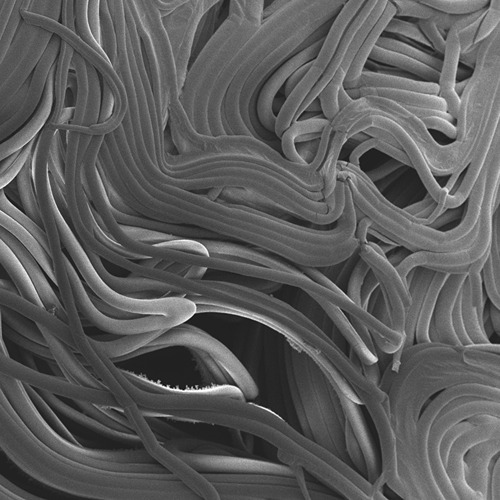
 Purdue University’s Daniel Szymanski developed a mass spectrometry method to identify the composition of protein complexes. Proteins that co-purify are predicted interactors. The profile data are generated from extracts isolated from developing Arabidopsis leaves. (Purdue Agricultural Communication photo)
Purdue University’s Daniel Szymanski developed a mass spectrometry method to identify the composition of protein complexes. Proteins that co-purify are predicted interactors. The profile data are generated from extracts isolated from developing Arabidopsis leaves. (Purdue Agricultural Communication photo) Featured Stories

By learning how to cut through the hype and focus on what truly matters, you can shop smarter...

Channing Arndt, research professor in agricultural economics and director of the Global Trade...
Daniel Szymanski and his former postdoctoral scholar Youngwoo Lee, lead author on their paper...

The Costa Rica Rainforest Experience study abroad trip allowed students to experience the wonders...

Trask is writing a new narrative about rats, birds and people as he pursues his doctorate in...
Several Forestry and Natural Resources students, staff and recent alumni have been recognized for...